 Hafsa Tariq1
Hafsa Tariq1 Mirela Dobre2*
Mirela Dobre2*- 1Division of Nephrology, University of Rochester Medical Center, Rochester, NY, United States
- 2Division of Nephrology and Hypertension, University Hospitals Cleveland Medical Center, Case Western Reserve University, Cleveland, OH, United States
Metabolic acidosis, a common complication in patients with chronic kidney disease (CKD), results in a multitude of deleterious effects. Though the restoration of kidney function following transplantation is generally accompanied by a correction of metabolic acidosis, a subset of transplant recipients remains afflicted by this ailment and its subsequent morbidities. The vulnerability of kidney allografts to metabolic acidosis can be attributed to reasons similar to pathogenesis of acidosis in non-transplant CKD, and to transplant specific causes, including donor related, recipient related, immune mediated factors, and immunosuppressive medications. Correction of metabolic acidosis in kidney transplantation either with alkali therapy or through dietary manipulations may have potential benefits and the results of such clinical trials are eagerly awaited. This review summarizes the published evidence on the pathogenesis and clinical consequences of chronic metabolic acidosis in kidney transplant recipients.
Introduction
Metabolic acidosis, defined as serum total CO2 level below 22mEq/L is a common complication in advanced chronic kidney diseases (CKD), and there is a graded association with CKD severity (Dobre et al., 2013). Kidney transplantation, the preferred treatment modality for kidney replacement therapy is associated with restoration of several essential kidney functions, however acid base abnormalities are slow to correct. It is estimated that the prevalence of metabolic acidosis ranges from 12% to 58% (Park et al., 2017; Djamali et al., 2019) in kidney transplant recipients and it can occur at a relatively higher estimated glomerular filtration rate (eGFR) when compared to general population with non-transplant CKD. This is primarily related to the inability of the single functioning kidney to excrete the daily acid load, added to the tubular toxicity of the immunosuppressive medications, particularly calcineurin inhibitors. Recipient and donor characteristics have been postulated to also play a role in the development of mild to moderate degrees of chronic metabolic acidosis after kidney transplantation.
Persistent metabolic acidosis can cause several complications. These include, but are not limited to the development of metabolic bone disease, sarcopenia, insulin resistance, anemia, kidney disease progression, and increased all-cause mortality in individuals with non-transplant CKD (Dobre et al., 2013). It is reasonable to assume that the same consequences will be encountered in metabolic acidosis occurring after kidney transplantation, though the evidence is somewhat limited. The association with eGFR decline and allograft loss has been described (Park et al., 2017), as well as the association with cardiovascular morbidity and mortality, although the evidence remains scarce (Djamali et al., 2019; Bohling et al., 2021).
In this review, we describe the proposed mechanisms of the development of metabolic acidosis in kidney transplant recipients, and detail the factors pertinent to chronic kidney disease in general, and those specific to kidney transplantation. We also discuss the complications associated with chronic metabolic acidosis and the available evidence in support of its treatment in kidney transplant recipients.
Epidemiology
The actual prevalence of metabolic acidosis in kidney transplant recipients remains unclear, primarily due to multiple definitions being used and the timing relative to kidney transplantation event. Evidence for impaired tubular hydrogen ion transport, and diminished renal ammonia production was mechanistically studied and reported for the first time in 1967 in a transplant patient with multiple tubular defects (Massry et al., 1967). The presence of renal tubular acidosis alone has been described in multiple studies since then, most of the data obtained in the early post kidney transplantation period (Massry et al., 1967; Better et al., 1970; Wilson and Siddiqui, 1973). More recently, in cross-sectional studies, the incidence of renal tubular acidosis has been reported up to 33–35% of kidney transplant recipients (Keven et al., 2007; Malik et al., 2011).
In a cross sectional study of 823 individuals with a kidney transplant, 58% of participants had a sodium bicarbonate concentration less than 24 mmol/L (Yakupoglu et al., 2007) In a large retrospective cohort of 2318 kidney transplant recipients (Park et al., 2017), almost 14% of participants with eGFR 30–60 ml/min/1.73 m (Park et al., 2017), had metabolic acidosis at six months post-transplant, and this remained relatively persistent at 60 months post-transplant with 16% of participants having metabolic acidosis. Amongst the individuals with eGFR 15–30 ml/min/1.73 m (Park et al., 2017), the prevalence of metabolic acidosis was much higher, estimated at 63% at 6 months, and 60% at 60 months post-transplant. In a post hoc analysis of an open label randomized study of 90 kidney transplant recipients, the prevalence of metabolic acidosis was as high as 63% immediately after the transplantation. This decreased to 28% at 12 months post-transplant as the eGFR increased (Wiegand et al., 2019).
Pathophysiology
A healthy kidney maintains acid-base homeostasis primarily by ammoniagenesis, regeneration of bicarbonate and excretion of hydrogen ions (Kraut and Madias, 2016). As the kidney function is lost and replaced by the solitary kidney allograft, the amount of ammonia production per nephron increases as a compensatory mechanism. This gives rise to hyperchloremic (non-anion gap) metabolic acidosis. However, the excretion of urinary acid continues to decrease. The increase ammonia production leads to a vicious cycle involving intrarenal complement activation, which results in tubulointerstitial injury. This initiates a cascade of events reducing the kidney’s capacity to synthesize ammonia and further decreasing urinary acid excretion (Clark et al., 1990; Raphael et al., 2017; Raphael, 2018) (Figure 1). Accumulation of phosphate, sulfate, and other non-volatile acids takes place eventually leading to high anion gap metabolic acidosis, as the allograft function is progressively lost.
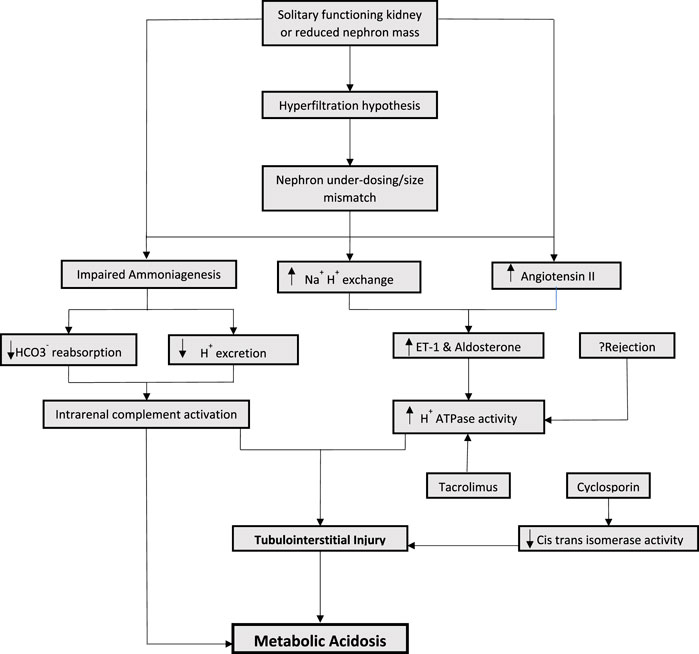
FIGURE 1. Mechanisms of metabolic acidosis in kidney transplantation.
Increased endogenous endothelin has been proposed to play a role in the progression of kidney disease in animal models (Wesson, 2001). Endothelin-1 (ET-1) is known to promote acid excretion by enhancing Na+/H+ exchange in proximal and distal tubules which reduces bicarbonate secretion and stimulates aldosterone. Consequently, the increase in aldosterone leads to an increase in H + ATPase activity, resulting in inflammation and tubulointerstitial injury (Wesson, 2001; Bento et al., 2007; Wesson et al., 2011; Wesson et al., 2015). In rat models who underwent subtotal nephrectomy, increased levels of systemic and renal endothelin-1 were observed (Wesson, 2001). Despite normal serum bicarbonate, intra renal acid accumulation and renal angiotensin II levels are increased which result in increased ET-1 and aldosterone levels, leading to tubular toxicity. It is important to note that angiotensin II receptor antagonism alone was not as effective for preserving eGFR decline compared to alkali supplementation, which resulted in decreasing intra renal acid accumulation (Wesson, 2001; Wesson et al., 2011; Wesson et al., 2015).
The mechanism of nephron hyperfiltration and compensatory increase in ammoniagenesis leading to complement activation and increased endothelin and aldosterone is especially important in kidney transplant recipients. Hyperfiltration hypothesis has been well described in kidney transplant recipients (Terasaki et al., 1994). By virtue of having a solitary functioning kidney and therefore, reduced nephron mass, the mechanisms of metabolic acidosis post kidney transplantation can be extrapolated from observations derived from animal models with subtotal nephrectomies. In addition to having a solitary kidney, factors pertinent to donor and recipient characteristics also play a significant role in nephron hyperfiltration (Keven et al., 2007; Kocyigit et al., 2010).
The concept of “nephron underdosing” was described by Brenner in 1993 as a potential cause for long term allograft failure (Brenner and Milford, 1993). It emphasized the importance of size mismatch between the donor and recipient. Hyperfiltration hypothesis was later described by Terasaki, demonstrating that reduced renal mass due to donor-recipient size mismatch results in compensatory nephron hypertrophy and nephron burn out resulting in chronic allograft failure (Terasaki et al., 1994). This is probably true for both living and deceased donor recipients. Reduced nephron mass can lead to proportionately reduced excretion of urinary acid, therefore contributing to development of acidosis.
Immunosuppressive medications, most notably, calcineurin inhibitors play an important role in the development of metabolic acidosis in transplant recipients. Unlike acidosis seen with nephron hypertrophy, that can be either non-anion gap or high anion gap acidosis, calcineurin inhibitors are typically associated with renal tubular acidosis (RTA) (Watanabe et al., 2005; Mohebbi et al., 2009). Calcineurin inhibitors (CNIs) are known to have a dose and duration dependent effect causing renal tubular toxicity. This tubular toxicity resulting in RTA is hypothesized to be more functional rather than structural. Hyperchloremic (non-anion gap) metabolic acidosis associated with CNI administration develops only in the setting of increased intrarenal post-glomerular blood flow and has been observed to be accompanied by the CNI arteriolopathy (Kaneko et al., 2022). Several mechanism of CNI induced hyperchloremic metabolic acidosis have been proposed in animal models, including impaired secretion of both H+ and K+, and downregulation of ammonia transporters in the collecting ducts (Kaneko et al., 2022). In humans, activation of the sodium- chloride cotransporter in the distal tubular cells leads to an impaired negative potential in the collecting duct lumen, and manifests clinically as hyperkalemic RTA and hypertension. Specific differences exist between the two most frequently used CNIs. Cyclosporin causes renal tubular acidosis by blocking the peptidyl prolyl cis-trans isomerase activity through a cyclophilin- dependent mechanism. Such inhibition may cause distal renal tubular acidosis (Watanabe et al., 2005). Tacrolimus, on the other hand, has been noted to affect major transport proteins that are involved in acid base homeostasis in the proximal and distal tubules, including endothelin 1 and H + - ATPase transport protein (Mohebbi et al., 2009). This dose dependent tubular toxicity may be reversed with reduction in the medication dose (Mohebbi et al., 2009).
Rejection can cause tubulitis and ischemic tubular dysfunction, affecting H + ATPase activity and anion exchanger leading to RTA (Mookerjee et al., 1969; Batlle et al., 1981). However, the data proving the association of rejection with metabolic acidosis is somewhat conflicting (Schwarz et al., 2006; Keven et al., 2007). There is reason to speculate that a feedback loop generated from local acidic environments can modulate immune responses, resulting in increased phagocytic activity of macrophages, thus enhancing inflammation (Riemann et al., 2016). Other medications used in the post-transplant period, including the antibiotic Trimethoprim—Sulfamethoxazole, can cause distal type 4 RTA (Pochineni and Rondon-Berrios, 2018).
Lastly, hyperkalemia, a common occurrence post kidney transplantation (generally related to medications, delayed graft function, potassium intake, etc) also contributes to development of acidosis. Hyperkalemia decreases ammonia generation and transport in proximal tubule and collecting duct respectively, leading to impaired ammonia excretion and subsequent metabolic acidosis (Harris et al., 2018). Of the post-transplant immunosuppressants, CNIs are particularly implicated in the development of hyperkalemia by suppression of mineralocorticoid receptor transcriptional activity, leading to signs of hypoaldosteronism (Deppe et al., 2002).
Complications of metabolic acidosis in kidney transplant recipients
Association with graft failure
Chronic metabolic acidosis that persists after kidney transplantation may be detrimental to allograft survival. In a large cohort of adult kidney transplant recipients followed for about 62 months (14,271.3 person-years in total), metabolic acidosis defined as the serum total CO2 level less than 22 mmol/L at three months post-transplant was associated with increased risk of graft loss and death censored graft failure (Park et al., 2017). Similar results were reported in other studies (Wiegand et al., 2019); however, others have failed to reproduce these findings (Schulte et al., 2019). This points towards the need for rigorously designed trials, focused on metabolic complications following kidney transplantation, as an adjunct to immunological therapies, with the ultimate goal to enhance graft and overall survival.
Anemia
Post transplantation anemia is common in kidney transplant patients with prevalence being reported around 30–50% of the patients (Yabu and Winkelmayer, 2011; Gafter-Gvili and Gafter, 2019). The pathogenesis appears to be multifactorial, including iron deficiency, reduced graft function, immunosuppressive medications, and erythropoietin resistance secondary to inflammatory state (Yabu and Winkelmayer, 2011). The correlation between metabolic acidosis and anemia has been shown in both hemodialysis and transplant patients (Yorgin et al., 2002). The exact mechanism remains unclear; however, it has been hypothesized that a rightward shift in the oxygen—hemoglobin dissociation curve resulting in downregulation of erythropoietin receptors could be one of the culprits (Ambuhl, 2007).
Bone disease
Metabolic bone disease after kidney transplantation is due to a host of pathophysiological processes and can take the form of either high or low bone turnover. Acidosis can reduce synthesis of 1,25 (OH)2 vitamin D3 by proximal tubule, increasing calcium excretion and serum PTH levels, thus promoting bone resorption (Jaeger et al., 1987; Alpern and Sakhaee, 1997; Yakupoglu et al., 2007; Zhang and Chouhan, 2012). In vitro acidic environment has been shown to increase resorptive activity of osteoclasts (Murrills et al., 1993). Additionally, pre-transplant bone disease may be worsened by the development of post-transplant acidosis and concomitant use of immunosuppressive regimen, especially corticosteroids and CNIs. As a consequence, close to 9% of bone mineral density may be lost at 18 months post-transplant (Julian et al., 1991), therefore increasing the risk of bone fractures in this patient population (Chiu et al., 1998).
Frailty and sarcopenia
Individuals with advanced kidney disease often have reduced muscle mass and exercise tolerance (Painter et al., 1986; Dobre et al., 2015; Dubey et al., 2020). Physical capacity and exercise tolerance increases only slightly after kidney transplantation, and this remains an active area of research. Metabolic acidosis has been shown to be one of the contributing factors to sarcopenia after kidney transplantation. Acidosis induces muscle catabolism and inhibits muscle protein synthesis (May et al., 1987; Bailey et al., 2006). It has also been associated with higher serum cortisol levels; the rate of protein catabolism is indirectly associated with serum bicarbonate levels and directly to serum cortisol levels (Garibotto et al., 1994). Correction of acidosis with serum bicarbonate supplementation in chronic kidney disease has shown to decrease proteolysis and improve muscle mass (Reaich et al., 1993; Dubey et al., 2020), but these outcomes have not been studied in kidney transplant recipients. In addition, post kidney transplant patients have been known to have hypophosphatemia. This is largely due to inappropriate renal excretion of phosphorus related to multiple factors including hyperparathyroidism, metabolic acidosis, and tubular dysfunction (Guntupalli et al., 1982; Ghanekar et al., 2006). Increased phosphaturia can result in depletion of intramuscular ATP stores, resulting in sarcopenia.
Mortality
Metabolic acidosis has been shown to be an independent risk factor for all-cause mortality in CKD (Navaneethan et al., 2011; Raphael et al., 2011; Raphael et al., 2016). These findings were replicated in kidney transplant population. In a single center observational study, comparing kidney transplant recipients with serum bicarbonate 24-25.9 mEq/L, those with serum bicarbonate levels less than 24 mEq/L at one year post transplantation had increased risk of cardiovascular events and all-cause mortality (Djamali et al., 2019). The direct role of metabolic acidosis on increased mortality remains unclear, however worsening cardiovascular disease, including altering the heart contractile function and lowering the trigger for arrhythmias in the setting of acidosis may explain this observed association.
Metabolic acidosis treatment in kidney transplant recipients
There is a paucity of evidence attesting to the benefits of metabolic acidosis correction in kidney transplant recipients. In fact, in a large retrospective study of 4741 kidney transplant recipients sodium bicarbonate therapy was associated with higher risk of graft failure (Schulte et al., 2019). However there were significant limitations of this study, including the reliance of health insurance data to define the study groups, and the lack of laboratory values to assess allograft function and acid base status.
In a small randomized controlled trial aimed to investigate the effect of sodium bicarbonate therapy on vascular endothelial function in 20 kidney transplant recipients (Bohling et al., 2021), the sodium bicarbonate therapy was considered safe and not associated with increased blood pressures, weight gain, or change in inflammatory markers. A larger trial, looking at similar outcomes in kidney transplant recipients is underway (NCT05005793). The results of Preserve—Transplant study, a multi-center randomized controlled trial designed to investigate the effect of sodium bicarbonate therapy in preserving kidney allograft function and slowing progression of chronic kidney disease in kidney transplant recipients over a period of two years (Wiegand et al., 2018) are eagerly awaited (NCT03102996).
Data on dietary fruits and vegetable consumption as a measure of metabolic acidosis correction in kidney transplant recipients is even scarcer. In CKD, a diet rich in fruits and vegetables favorably affects acid-base metabolism, neutralizes diet-induced acid, and has been shown to be cardio-protective (Goraya et al., 2021). In a prospective cohort of 400 kidney transplant recipients, consumption of a vegetable enriched diet was associated with lower cardiovascular and all-cause mortality (Sotomayor et al., 2020). We propose that the reduced dietary acid load directly derived from a high fruits and vegetables diet contributes to the reduced cardiovascular risk, however the acid base parameters pre- and post-therapy were not reported (Sotomayor et al., 2020).
Conclusion
Metabolic acidosis is an understudied, highly prevalent complication in kidney transplant recipients. In addition to mechanisms described in metabolic acidosis of CKD, calcineurin inhibitors play a central role in its development and maintenance, through dysregulation of tubular transport proteins involved in acid load handling. Additional research aimed to unfold the mechanisms and consequences of metabolic acidosis present post kidney transplantation will help inform future large intervention trials designed to correct it, in an attempt to prevent allograft loss and improve overall survival in kidney transplant recipients.
Author contributions
HT contributed to the concept, interpretation of data, and drafted the original manuscript. MD contributed to the concept, interpretation of data, and critically revised it for important intellectual content. Both authors approved the final version of the manuscript.
Funding
NIH/NHLBI 5R01HL141846.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alpern R. J., Sakhaee K. (1997). The clinical spectrum of chronic metabolic acidosis: homeostatic mechanisms produce significant morbidity. Am. J. Kidney Dis. 29 (2), 291–302. doi:10.1016/s0272-6386(97)90045-7
Ambuhl P. M. (2007). Posttransplant metabolic acidosis: a neglected factor in renal transplantation? Curr. Opin. Nephrol. Hypertens. 16 (4), 379–387. doi:10.1097/MNH.0b013e3281bd8860
Bailey J. L., Zheng B., Hu Z., Price S. R., Mitch W. E. (2006). Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J. Am. Soc. Nephrol. 17 (5), 1388–1394. doi:10.1681/ASN.2004100842
Batlle D. C., Mozes M. F., Manaligod J., Arruda J. A., Kurtzman N. A. (1981). The pathogenesis of hyperchloremic metabolic acidosis associated with kidney transplantation. Am. J. Med. 70 (4), 786–796. doi:10.1016/0002-9343(81)90534-9
Bento L. M., Fagian M. M., Vercesi A. E., Gontijo J. A. (2007). Effects of NH4Cl-induced systemic metabolic acidosis on kidney mitochondrial coupling and calcium transport in rats. Nephrol. Dial. Transpl. 22 (10), 2817–2823. doi:10.1093/ndt/gfm306
Better O. S., Chaimowitz C., Alroy G. G., Sisman I. (1970). Spontaneous remission of the defect in urinary acidification after cadaver kidney homotransplantation. Lancet 1 (7638), 110–112. doi:10.1016/s0140-6736(70)90464-2
Bohling R., Grafals M., Moreau K., You Z., Tommerdahl K. L., Bjornstad P., et al. (2021). A pilot study of the safety and efficacy of alkali therapy on vascular function in kidney transplant recipients. Kidney Int. Rep. 6 (9), 2323–2330. doi:10.1016/j.ekir.2021.06.006
Brenner B. M., Milford E. L. (1993). Nephron underdosing: a programmed cause of chronic renal allograft failure. Am. J. Kidney Dis. 21 (2), 66–72. doi:10.1016/0272-6386(93)70097-i
Chiu M. Y., Sprague S. M., Bruce D. S., Woodle E. S., Thistlethwaite J. R., Josephson M. A. (1998). Analysis of fracture prevalence in kidney-pancreas allograft recipients. J. Am. Soc. Nephrol. 9 (4), 677–683. doi:10.1681/ASN.V94677
Clark E. C., Nath K. A., Hostetter M. K., Hostetter T. H. (1990). Role of ammonia in tubulointerstitial injury. Min. Electrolyte Metab. 16 (5), 315–321.
Deppe C. E., Heering P. J., Viengchareun S., Grabensee B., Farman N., Lombes M. (2002). Cyclosporine a and FK506 inhibit transcriptional activity of the human mineralocorticoid receptor: a cell-based model to investigate partial aldosterone resistance in kidney transplantation. Endocrinology 143 (5), 1932–1941. doi:10.1210/endo.143.5.8821
Djamali A., Singh T., Melamed M. L., Stein J. H., Aziz F., Parajuli S., et al. (2019). Metabolic acidosis 1 Year following kidney transplantation and subsequent cardiovascular events and mortality: An observational cohort study. Am. J. Kidney Dis. 73 (4), 476–485. doi:10.1053/j.ajkd.2018.12.023
Dobre M., Rahman M., Hostetter T. H. (2015). Current status of bicarbonate in CKD. J. Am. Soc. Nephrol. 26 (3), 515–523. doi:10.1681/ASN.2014020205
Dobre M., Yang W., Chen J., Drawz P., Hamm L. L., Horwitz E., et al. (2013). Association of serum bicarbonate with risk of renal and cardiovascular outcomes in CKD: a report from the chronic renal insufficiency cohort (CRIC) study. Am. J. Kidney Dis. 62 (4), 670–678. doi:10.1053/j.ajkd.2013.01.017
Dubey A. K., Sahoo J., Vairappan B., Haridasan S., Parameswaran S., Priyamvada P. S. (2020). Correction of metabolic acidosis improves muscle mass and renal function in chronic kidney disease stages 3 and 4: a randomized controlled trial. Nephrol. Dial. Transpl. 35 (1), 121–129. doi:10.1093/ndt/gfy214
Gafter-Gvili A., Gafter U. (2019). Posttransplantation anemia in kidney transplant recipients. Acta Haematol. 142 (1), 37–43. doi:10.1159/000496140
Garibotto G., Russo R., Sofia A., Robaudo C., Sala M. R., Moscatelli P., et al. (1994). Skeletal muscle protein synthesis and degradation in patients with chronic renal failure. Kidney Int. 45 (5), 1432–1439. doi:10.1038/ki.1994.187
Ghanekar H., Welch B. J., Moe O. W., Sakhaee K. (2006). Post-renal transplantation hypophosphatemia: a review and novel insights. Curr. Opin. Nephrol. Hypertens. 15 (2), 97–104. doi:10.1097/01.mnh.0000203187.49890.cc
Goraya N., Munoz-Maldonado Y., Simoni J., Wesson D. E. (2021). Treatment of chronic kidney disease-related metabolic acidosis with fruits and vegetables compared to NaHCO3 yields more and better overall health outcomes and at comparable five-year cost. J. Ren. Nutr. 31 (3), 239–247. doi:10.1053/j.jrn.2020.08.001
Guntupalli J., Eby B., Lau K. (1982). Mechanism for the phosphaturia of NH4Cl: dependence on acidemia but not on diet PO4 or PTH. Am. J. Physiol. 242 (5), F552–F560. doi:10.1152/ajprenal.1982.242.5.F552
Harris A. N., Grimm P. R., Lee H. W., Delpire E., Fang L., Verlander J. W., et al. (2018). Mechanism of hyperkalemia-induced metabolic acidosis. J. Am. Soc. Nephrol. 29 (5), 1411–1425. doi:10.1681/ASN.2017111163
Jaeger P., Jones W., Kashgarian M., Segre G. V., Hayslett J. P. (1987). Parathyroid hormone directly inhibits tubular reabsorption of bicarbonate in normocalcaemic rats with chronic hyperparathyroidism. Eur. J. Clin. Invest. 17 (5), 415–420. doi:10.1111/j.1365-2362.1987.tb01136.x
Julian B. A., Laskow D. A., Dubovsky J., Dubovsky E. V., Curtis J. J., Quarles L. D. (1991). Rapid loss of vertebral mineral density after renal transplantation. N. Engl. J. Med. 325 (8), 544–550. doi:10.1056/NEJM199108223250804
Kaneko S., Usui J., Takahashi K., Oda T., Yamagata K. (2022). Increased intrarenal post-glomerular blood flow is a key condition for the development of calcineurin inhibitor-induced renal tubular acidosis in kidney transplant recipients. Clin. Transpl. 36 (6), e14648. doi:10.1111/ctr.14648
Keven K., Ozturk R., Sengul S., Kutlay S., Ergun I., Erturk S., et al. (2007). Renal tubular acidosis after kidney transplantation--incidence, risk factors and clinical implications. Nephrol. Dial. Transpl. 22 (3), 906–910. doi:10.1093/ndt/gfl714
Kocyigit I., Unal A., Kavuncuoglu F., Sipahioglu M. H., Tokgoz B., Oymak O., et al. (2010). Renal tubular acidosis in renal transplantation recipients. Ren. Fail. 32 (6), 687–690. doi:10.3109/0886022X.2010.486494
Kraut J. A., Madias N. E. (2016). Metabolic acidosis of CKD: An update. Am. J. Kidney Dis. 67 (2), 307–317. doi:10.1053/j.ajkd.2015.08.028
Malik S. I., Naqvi R., Ahmed E., Zafar M. N. (2011). Prevalence and risk factors of renal tubular acidosis after kidney transplantation. J. Pak. Med. Assoc. 61 (1), 23–27.
Massry S. G., Preuss H. G., Maher J. F., Schreiner G. E. (1967). Renal tubular acidosis after cadaver kidney homotransplantation. Studies on mechanism. Am. J. Med. 42 (2), 284–292. doi:10.1016/0002-9343(67)90026-5
May R. C., Kelly R. A., Mitch W. E. (1987). Mechanisms for defects in muscle protein metabolism in rats with chronic uremia. Influence of metabolic acidosis. J. Clin. Invest. 79 (4), 1099–1103. doi:10.1172/JCI112924
Mohebbi N., Mihailova M., Wagner C. A. (2009). The calcineurin inhibitor FK506 (tacrolimus) is associated with transient metabolic acidosis and altered expression of renal acid-base transport proteins. Am. J. Physiol. Ren. Physiol. 297 (2), F499–F509. doi:10.1152/ajprenal.90489.2008
Mookerjee B., Gault M. H., Dossetor J. B. (1969). Hyperchloremic acidosis in early diagnosis of renal allograft rejection. Ann. Intern. Med. 71 (1), 47–58. doi:10.7326/0003-4819-71-1-47
Murrills R. J., Stein L. S., Dempster D. W. (1993). Stimulation of bone resorption and osteoclast clear zone formation by low pH: a time-course study. J. Cell. Physiol. 154 (3), 511–518. doi:10.1002/jcp.1041540309
Navaneethan S. D., Schold J. D., Arrigain S., Jolly S. E., Wehbe E., Raina R., et al. (2011). Serum bicarbonate and mortality in stage 3 and stage 4 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 6 (10), 2395–2402. doi:10.2215/CJN.03730411
Painter P., Messer-Rehak D., Hanson P., Zimmerman S. W., Glass N. R. (1986). Exercise capacity in hemodialysis, CAPD, and renal transplant patients. Nephron 42 (1), 47–51. doi:10.1159/000183632
Park S., Kang E., Park S., Kim Y. C., Han S. S., Ha J., et al. (2017). Metabolic acidosis and long-term clinical outcomes in kidney transplant recipients. J. Am. Soc. Nephrol. 28 (6), 1886–1897. doi:10.1681/ASN.2016070793
Pochineni V., Rondon-Berrios H. (2018). Electrolyte and acid-base disorders in the renal transplant recipient. Front. Med. 5, 261. doi:10.3389/fmed.2018.00261
Raphael K. L., Carroll D. J., Murray J., Greene T., Beddhu S. (2017). Urine ammonium predicts clinical outcomes in hypertensive kidney disease. J. Am. Soc. Nephrol. 28 (8), 2483–2490. doi:10.1681/ASN.2016101151
Raphael K. L. (2018). Metabolic acidosis and subclinical metabolic acidosis in CKD. J. Am. Soc. Nephrol. 29 (2), 376–382. doi:10.1681/ASN.2017040422
Raphael K. L., Murphy R. A., Shlipak M. G., Satterfield S., Huston H. K., Sebastian A., et al. (2016). Bicarbonate concentration, acid-base status, and mortality in the health, aging, and body composition study. Clin. J. Am. Soc. Nephrol. 11 (2), 308–316. doi:10.2215/CJN.06200615
Raphael K. L., Wei G., Baird B. C., Greene T., Beddhu S. (2011). Higher serum bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kidney Int. 79 (3), 356–362. doi:10.1038/ki.2010.388
Reaich D., Channon S. M., Scrimgeour C. M., Daley S. E., Wilkinson R., Goodship T. H. (1993). Correction of acidosis in humans with CRF decreases protein degradation and amino acid oxidation. Am. J. Physiol. 265 (1), E230–E235. doi:10.1152/ajpendo.1993.265.2.E230
Riemann A., Wussling H., Loppnow H., Fu H., Reime S., Thews O. (2016). Acidosis differently modulates the inflammatory program in monocytes and macrophages. Biochim. Biophys. Acta 1862 (1), 72–81. doi:10.1016/j.bbadis.2015.10.017
Schulte K., Puchel J., Schussel K., Borzikowsky C., Kunzendorf U., Feldkamp T. (2019). Effect of sodium bicarbonate in kidney transplant recipients with chronic metabolic acidosis. Transpl. Direct 5 (7), e464. doi:10.1097/TXD.0000000000000910
Schwarz C., Benesch T., Kodras K., Oberbauer R., Haas M. (2006). Complete renal tubular acidosis late after kidney transplantation. Nephrol. Dial. Transpl. 21 (9), 2615–2620. doi:10.1093/ndt/gfl211
Sotomayor C. G., Gomes-Neto A. W., Eisenga M. F., Nolte I. M., Anderson J. L. C., de Borst M. H., et al. (2020). Consumption of fruits and vegetables and cardiovascular mortality in renal transplant recipients: a prospective cohort study. Nephrol. Dial. Transpl. 35 (2), 357–365. doi:10.1093/ndt/gfy248
Terasaki P. I., Koyama H., Cecka J. M., Gjertson D. W. (1994). The hyperfiltration hypothesis in human renal transplantation. Transplantation 57 (10), 1450–1454. doi:10.1097/00007890-199405000-00008
Watanabe S., Tsuruoka S., Vijayakumar S., Fischer G., Zhang Y., Fujimura A., et al. (2005). Cyclosporin A produces distal renal tubular acidosis by blocking peptidyl prolyl cis-trans isomerase activity of cyclophilin. Am. J. Physiol. Ren. Physiol. 288 (1), F40–F47. doi:10.1152/ajprenal.00218.2004
Wesson D. E. (2001). Endogenous endothelins mediate increased acidification in remnant kidneys. J. Am. Soc. Nephrol. 12 (9), 1826–1835. doi:10.1681/ASN.V1291826
Wesson D. E., Jo C. H., Simoni J. (2015). Angiotensin II-mediated GFR decline in subtotal nephrectomy is due to acid retention associated with reduced GFR. Nephrol. Dial. Transpl. 30 (5), 762–770. doi:10.1093/ndt/gfu388
Wesson D. E., Simoni J., Broglio K., Sheather S. (2011). Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am. J. Physiol. Ren. Physiol. 300 (4), F830–F837. doi:10.1152/ajprenal.00587.2010
Wiegand A., Graf N., Bonani M., Frey D., Wuthrich R. P., Mohebbi N. (2019). Relationship of serum bicarbonate levels with 1-year graft function in kidney transplant recipients in Switzerland. Kidney Blood Press. Res. 44 (5), 1179–1188. doi:10.1159/000502527
Wiegand A., Ritter A., Graf N., Arampatzis S., Sidler D., Hadaya K., et al. (2018). Preservation of kidney function in kidney transplant recipients by alkali therapy (Preserve-Transplant study): rationale and study protocol. BMC Nephrol. 19 (1), 177. doi:10.1186/s12882-018-0956-8
Wilson D. R., Siddiqui A. A. (1973). Renal tubular acidosis after kidney transplantation. Natural history and significance. Ann. Intern. Med. 79 (3), 352–361. doi:10.7326/0003-4819-79-3-352
Yabu J. M., Winkelmayer W. C. (2011). Posttransplantation anemia: mechanisms and management. Clin. J. Am. Soc. Nephrol. 6 (7), 1794–1801. doi:10.2215/CJN.01190211
Yakupoglu H. Y., Corsenca A., Wahl P., Wuthrich R. P., Ambuhl P. M. (2007). Posttransplant acidosis and associated disorders of mineral metabolism in patients with a renal graft. Transplantation 84 (9), 1151–1157. doi:10.1097/01.tp.0000287430.19960.0e
Yorgin P. D., Scandling J. D., Belson A., Sanchez J., Alexander S. R., Andreoni K. A. (2002). Late post-transplant anemia in adult renal transplant recipients. An under-recognized problem? Am. J. Transpl. 2 (5), 429–435. doi:10.1034/j.1600-6143.2002.20506.x
Keywords: metabolic acidosis, kidney transplant, renal tubular acidosis, tubulointerstial injury, graft failure
Citation: Tariq H and Dobre M (2022) Metabolic acidosis post kidney transplantation. Front. Physiol. 13:989816. doi: 10.3389/fphys.2022.989816
Received: 08 July 2022; Accepted: 01 August 2022;
Published: 23 August 2022.
Edited by:
Snezana Petrovic, Campbell University, United StatesReviewed by:
Pierre-Yves Martin, Hôpitaux Universitaires de Genève, SwitzerlandCopyright © 2022 Tariq and Dobre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirela Dobre, mirela.dobre@uhhospitals.org